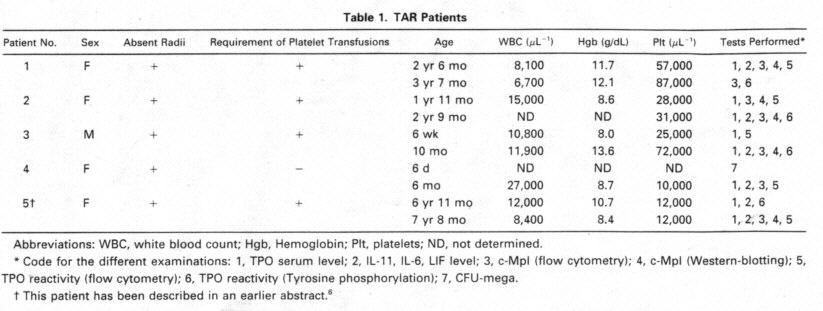
short ulnae, as weil as a
severe thrombocytopenia. lead to the diagno- sis of TAR. Megakaryoeytes were
completely absent in a bone mar row sample from the first week of life. This
sample was used for the clonogenic assay presented here.
Patient 5, a 7-year old
girl, has been described in an earlier publi- cation by us.6 She
showed absent radii and short u1nae~ thumb hypo- plasia and shoulder-girdle
involvement. in addition, a VSD was stated. In contrast with other reports about
TAR-syndrome. bleeding problems continued up to now. For instance, in the last
vear she had problems with petechiae and bleeding w ith platelet counts between
10,000 and 2O,OOO /µL.
Matenais. rhTPO
was provided by Dr A. Shimosaka Kinn Brewery (Tokyo, Japan). rh granulocyte
colony-stimulating factor (G-CSF) and rh stern cell factor (SCF) were provided
by Amgen (Thousand Oaks, CA). rh granulocyte-macrophage colony-stimuiat ing
tactor (GM-CSF) and rh interleukin-3 (IL-3) were a gift frorn Behringwerke
(Marburg, Germany), rh erythmpoietin (EPO) was obtained from Boehringer Mannheim
(Mannheim Germanv). Aden osine diphosphate (ADP), prostaglandin El (PGEI),
acetyl salicvlic acid, apyrase (type VIII), and bovine serum albumine (BSA) were
purchased from Sigma (Deisenhofen, Germany). The thrombin re ceptor agonist
peptide (serine-phenylaiani ne-leucine-ieucine-arginine-asparagine, TRAP) was
purchased from Bachem (Heidelberg Gerrnany). Monodonal anti bodies against CD62
and CD4 1, as weil as the IgG isotype control were purchased ftom Immunotech
(Ham burg, Germany), the monodonal antibody against human c-Vlpl was obtained
from Genzyme (Rüsselsheim Germany), the horse-radish peroxidase (HRP)-conjugated
recombinant antiphosphotyrosine anti body RC2O was purchased from Transduction
Laboratories (Lexing ton, KY). The reagents for the enhanced chemiluminescence (ECL)
and the 3H-thymidine were obtained from Amersham (Braun schweig,
Germany). Ccli culture media and fetal calf serum were purchased from Life
Technologies (Eggenstein, Germany).
TPO serum leveis. TPO
serum leveis were measured in a bioas say using the murine IL-3-dependent 32D (elone
23) cells24 trans fected with the human c-mpl.'2 Serum
samples were preabsorbed with the parental 32D (done 23) cells to remove
compiement activ ity. For the assay, 32D/MpI ceils were washed free of IL-3 and
plated (3,000 ceils, 100 µL total volume per weil) in flat-bottomed 96-weil
piates (Nunc, Wiesbaden, Germany:) in RPMI 1640 medium suppiemented with 5%
heat-inactivated fetal caif serum in 100 µL serial dilutions of test sera in
assay medium. After 72 hours, the microtiter cultures were pulsed with 3H-thymidine
(0.5 ,iCi/weil, specific activity 5 Ci/mmol) for another 4 hours. The
radioactive uptake was determined in a liquid scintillation counter. Serial dilu
tions of rhTPO in preabsorbed human serum were used as standards
the concentrations of the
samples were calcuiated from the standard curve by probit analysis. The
sensitivity of the assay was 100 pg/ mL.
Senirn leve/s of JL-6,
IL-Il, aiid leitkeinia inhibitorvfactoi (LIF). Serum
levels of IL-6 and IL- 11 were measured in commerciaily available enzyme-linked
immunosorbent assay (ELISA) systems (Quantikine. R&D systems, Abingdon. UK).
Detection iimits were 8 pg/mL for IL 6 and 30 pg/mL for IL 11 Serum leveis ot
LIF were measured in a sandwich ELISA.²5 Briefly, a monodonal antibody (MoAb)
against LIF served tor capturin a polvclonal rabbit antise rum was used tor
detection. The detection limit of this assay was 30 pg/mL.
Assay for CFUs. Bone
marrow mononudear edis (BM-MNCs, 105) obtained by densitv gradient
centrifugation with Ficoii were cultured in dup1icates in 1 mL aliquots in a
semi solid medium containing 0 7% methyl cellulose (vlethocel A4C WAK
Chemie Bad Homburo Germanv), 30% human tresh trozen plasma (Blood Bank Medica1
Schoo1 Hannover Germany), 0.5 x 10-9) mol/L 2 mercaptoethanol in Iscove's
modified Dulbecco's medium in 35- mm culture dishes. Hematopoietic growth
factors were added as specified in the Results section. The cultures were
incubated at 370C in an atmosphere of 5% C02 and 100%
humidity for 14 days. After this time, colonies were analyzed and counted in an
inversion micro scope. Megakaryocytic colonies were picked, spinned on sudes,
and May-GrünwaldlGiemsa stained for verifying the megakaryocytic morphology of
the cells.
Costiinukition of'
J)l(1teiets with TPO. Blood of heaithy volun teers or TAR patients was obtained from an
antecubital vein through a 19-gauge needle with oniy a light toumiquet into a
piastic syringe containing trisodium citrate (10 mmol/L final concentration).
Stimu lation experiments were started within 15 minutes after blood collec tion.
Experiments were usually performed with whole blood, and in some cases (platelet
counts < 50,000/µL) we used platelet-rich plasma (PRP). For preparing PRP,
whoie blood was centrifuged at 200g for 20 minutes and the supematant PRP was
removed. All incubations were done at 370C. Five to 10 µL aliquots
of blood or PRP containing2 X 105 to 2 x 106 piatelets were added to
polysty rene tubes containing 60 µL of phosphate-buffered sahne (PBS; 130
mmol/L NaCI, 10 mmol/L sodium phosphate, pH 7.5) with various concentrations of
rhTPO (20 nglmL, unless indicated otherwise). After a preineubation of 5 minutes,
the platelet activators ADP (final concentration 50 µmol/L) or TRAP26 (final
concentration 5 µmol/ L) were added. The stimulation was stopped by addition of
1 rnL of a solution of 1 % formaldehyde in PBS. Unstimulated platelets serving
as a negative control were fixed immediately after blood collection or
preparation of PRP, respectively. The samples were
614 BALLMAIER ET AL
stored for 30 minutes on
ice before they were stained for flow cytometric analysis.
Flow cytomnetry. Fluorescei
n isothiocyanate (FITC)-conj ugated MoAb anti P-selectin (CD62; clone CLB-Thromb/6)
as anactiva tion-dependent antibody and phycoerythrine (PE)-conjugated anti
gplIbIIIIa (CD4 1; clone P2) as a pan-platelet marker were used for
determination of platelet activation. FITC- and PE-labeled isotype control
antibodies (Immunotech, Hamburg, Germany) were used as control. Fixed platelets
were pelleted (5 minutes, 2,00g), washed two times in FACS-buffer (PBS
supplemented with 0.1 % BSA and 0,1 % sodium azide and then resuspended in 40 µL
of a diluted human Ig solution (10 mg/mL GammaGard; Baxter. Un terschleißheim,
Germany) for blocking the Fc-receptors. After a short preincubation 10 µL of
each antibody solution was added followed by a 20-minute incubation on ice. To
determine the c-Mpl expression on patients' platelets we used an anti-c-Mpl MoAb
(vi 1.17 Genzyme): Piatelets were fixed and preincubated with human Ig as
described before. Cells were incubated with the M 1 antibody (10 µL in the
Ig-solution) or a nonspecific isotype control antibody for 30 minutes on ice.
Cells were washed in FACS-buffer and then incubated with FITC-conjugated rabbit
antimouse Ig (Dako, Glostrup, Denmark) according to manufacturer' s instructions.
After staining, platelets
were washed two times in FACS-buffer and analyzed on a FACScan flow cytometer (Becton-Dickinson).
FITC ~uorescence was detected with a 530/30 nm and PE fluores cence was detected
with a 585/42 nm band pass filter. Light scatter and fluorescence data were
obtained with gain settings in the ioga rithmic mode. Platelets were
distinguished from other cells, debris, and machine noise" on the basis of
their scatter profile. In some cases a fluorescence threshold was set to analyze
oniy those blood cells tliat had bound the PE-conjugated anti-CD4 1 antibody.
Platelet preparation
for gel electrophoresis and ilnmliloprecipitation. PRP
from whole blood was prepared as described above. PRP was incubated with acetyl
salicylic acid (2 mmol/L) for 30 minutes at room temperature. Then PGE 1 (1 µmol/L)
was added from a 1- mmol/L stock solution in absolute ethanol. A soft pellet was
obtained by centrifugation of the PRP at 800g for 10 minutes. The peilet was
resuspended in a modified HEPES-Tyrode buffer as recently described21 also
containing apyrase (2 U/mL) to avoid stimulation of platelets by ADP and washed
only once to minimize additional physical stress. The platelets were resuspended
in the same buffer, which was recalcified containing 1 mmoi/L CaCi2 at
370C.
Gel eiectrophoresis and
Western blotting. The
stimulation of platelets was terminated by adding an equal volume of 2x-concen-
trated sample buffer (10% giycerol, 1 % sodium dodecyi sulfate [SDS], 5%
2-mercapto-ethanol, 50 mmol/L Tris-HC1 pH 6.8, 10 mmolIL EGTA and 1 mmol/L
sodium orthovanadate and 0.002% bromophenol blue). SDS-gel electrophoresis of
platelet proteins was run on a 7.5% polyacrylamide gel after boiling the samples
for 5 minutes.
The proteins were
transferred onto a nitrocelluiose membrane by semidry Western blotting with a
buffer containing 50 mmol/L Tris, 40 mniol/L glycine, 0.04% SDS, and 17.5% methanol
for 1.5 hours at room temperature.
For blocking unoccupied
binding sites the membrane was incu bated in PBS-T (0.1% Tween 20 in PBS) with
1% BSA (blocking buffer). For the detection of phosphotyrosine or c-Mpl the
membrane was incubated in blocking buffer with MoAb against c-Mpl (done M
1,1:1,000) or with HRP-conjugated recombinant MoAb against phosphotyrosine
(RC2O, 1:2 ,500) overnight at 40C with gentle motion.
In the case of c-Mpl the
MoAb was removed and the membrane was washed four times with PBS-T and incubated
with HRP-conju gated second antibody (dii uted 1:1,000 in PBS-T) for 1 hour at
room temperature with gentle motion.
After washing the blots in
PBS-T four times the antibody reactions were detected by chemiluminescence with
the ECL reagents (Amer sham) according to the manufacturer' s instructions.
hnmunoprecipitation. The
platelet stimulation was terminated by adding an equal amount of lysis buffer
[15 mmol/L HEPES, 150 mmolIL NaC1, 1 mmol/L phenylmethylsulfonyl fluoride (PVISF),
10 mmol/L EGTA, 1 mmol/L sodium orthovanadate 0.8 mg/mL leupeptin, 2% (vol/vol)
Triton X-100, pH 7.4). The samples were incubate d for 20 to 30 minutes on ice
and atterwards the debns was pelleted at l0,OOOg for 20 minutes at 40C.
The supernatant was removed and precieared by adding 50 µL of 50% slurry of
protein A-agarose-beads (Upstate Biotechnology, New York. NY) for 1 liour in an
orbital shaker at 40C. The beads were centrifuged at 10,000g for 10
minutes, and the supematant was transferred into a new tube. The MoAb against
human c-Mpl (Genzyme) was added and incu bated for 4 hours or overnight in an
orbital shaker at 40C. Then the complex was captured adding protein
A-agarose-beads (50 µL of 50% slurry) for another 2 to 4 hours.
The immune complexes were
washed three times with ice-cooled immunoprecipitation-buffer (50 mmol/L Tris-CI:
pH 7.4;1% Noni det P-40; 150 mmol/L NaCT; 1 mmol/L EGTA; 1 nimoliL PMSF:
1 µg/mL of the protease
inhibitors aprotinin. leupeptin, and pep statin; 1 mmol/L sodium vanadate; and 1
mmol/L sodium fluoride). resuspended in an equal volume of 2X-sample buffer, and
detected by Western biotting after SDS-PAGE.
RESULTS
Senim leveis of TPO,
IL-6, IL-I1, and LIF. In healthy control persons TPO serum leveis normally were not detect able
in the bioassay (detection Jimit: 100 pg/mL). In contrast, TPO serum levels were
above the detection limit in all serum samples from TAR patients with the
exception of the first serum sample of patient 2 (Table 2).
Leveis of other cytokines
known to influence thrombocy topoiesis were evaluated in the sera of the TAR
patients. Serum levels of IL-6 and LIF were undetectable in all cases (detection
limits 8 respectively 30 pg /mL). In contrast IL 1 1 serum levels were elevated
above 300 pglmL in three out of five TAR patients (Table 2).
CFU-mega assay. Colony
forming assays were per formed with bone marrow cells of one patient only (no.
4). We had no consent of the other patients' parents for bone marrow evaluation
and there was no clinical need for bone marrow evaluation. In the patient tested,
no megakaryocytic colonies were observed after stimulation of BM-MNC with rhTPO
(10 ng/mL; Table 3). For comparison, in heaithy controls tlie median number of
megakaryocytic colonies was 11 (n = 12). In contrast, the numbers of CFU-GM and
BFU E colonies grown from the TAR patients' bone marrow were elevated as
compared with healthy controls (Table 3;).
Expression of c-Mpl on
piatelets. Expression
of the TPO receptor c-Mpl on platelets was determined by flow cytome try in all
patients and by Western blotting in four out of five TAR patients (patients no.
1, 2, 3, and 5) (Fig 1). Because the receptor is presumably expressed on all
cells of the mega karyocytic lineage, we used platelets as an example for c Mpl
expression in TAR patients. We could show a specific binding of a MoAb against
the c-Mpl on platelets of all patients tested. Fluorescence intensity after
staining with the anti-c-Mpl in platelets from the TAR patients was in the range
of normal controls (Fig lA). The molecular weight of